The Merajver research group is devoted to understanding the molecular genetics and associated functions of very aggressive breast cancer types. The primary areas of focus are systems biology, mathematical oncology, biophysics, cell biology, genetics, and drug development. A summary of the research currently underway in the lab is provided below.
It is widely known that cancer cells have altered metabolic characteristics when compared to normal cells. These adaptations enable the cancer cells to survive and potentially thrive in the nutrient-depleted environment that often results from tumor formation. The Merajver lab is studying the cellular energy balance in different cell lines that provide an in vitro model of tumor progression. We are looking for metabolic changes that correlate with the switch between cell proliferation and motility. This switch is expected to be important as it relates to primary tumor formation, where rapid proliferation is crucial, and to secondary metastasis, where motility to new sites is essential. In order to learn more about the switch from proliferation to motility, we are studying a wide variety of key metabolic pathways, as well as a variety of important motility regulators in these cells. Identifying the molecular and metabolic changes that promote metastasis may lead to the development of new drugs that prevent disease progression.
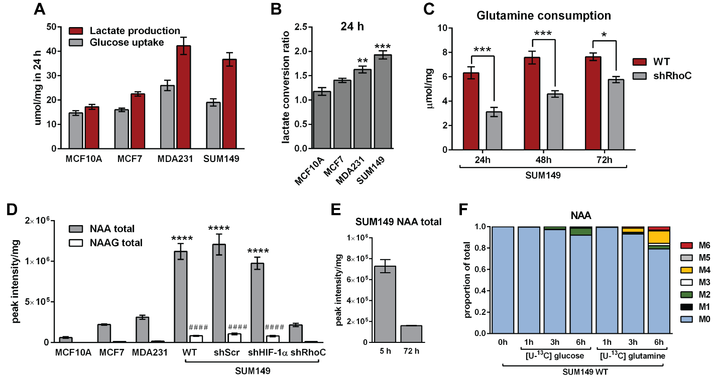
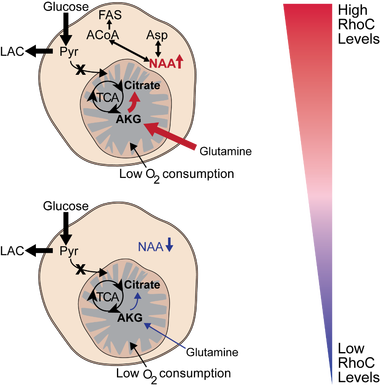
Recent work in the lab found that triple negative breast cancer cell lines MDA-MB-231 and SUM-149 convert a larger proportion of the glucose they consume to lactate (A), with the SUM-149 inflammatory breast cancer cell line exhibiting maximal glycolytic turnover of glucose to lactate (B). Indeed, SUM-149 cells are very glycolytic and rely on non-glucose carbons sources for TCA cycle function. To that end, we showed that these cells reductively carboxylate glutamine to citrate under atmospheric oxygen conditions. Furthermore, we identified a novel role for the pro-metastatic small GTPase RhoC in the metabolic reprogramming of inflammatory breast cancer cells. When RhoC is depleted in SUM-149 cells, glutamine uptake is significantly decreased (C). Additionally, we found that SUM-149 cells contain very high levels of the metabolite N-acetly-aspartate (NAA) when compared to other breast cancer and normal-like cell lines. Interestingly, when RhoC is depleted, the cells exhibit a drastic reduction in NAA levels (D). Importantly, we have demonstrated that the NAA pool is in a dynamic state, as the cells show reduced NAA levels when extracellular nutrients are depleted (E) and can be synthesized using extracellular glutamine, or to a lesser extent, glucose (F). Continued work to elucidate the role for NAA in inflammatory breast cancer is ongoing, as is work to identify mechanistic and signaling roles for RhoC in the metabolic adaptions in these extremely aggressive cancer cells.
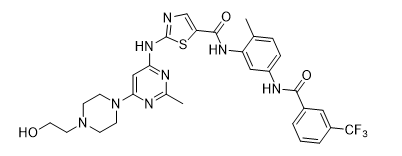
Triple-negative breast cancer (TNBC) is the only subset of breast cancers for which there are no FDA-approved targeted therapies. Despite advances in chemotherapy, the response rate for patients with TNBC is typically low, and the residual risk of recurrence is high. We recently developed a dual c-Src/p38 kinase inhibitor, UM-164, which potently inhibits the kinases, c-Src, p38α, and p38β. UM-164 demonstrates significant anti-TNBC activity both in vitro and in vivo. In contrast with FDA-approved inhibitors, dasatinib and bosutinib, which inhibit c-Src but not p38, UM-164 is observed to be significantly more efficacious in xenograft models of TNBC. Ongoing work involves the development of analogs of UM-164 optimized for oral absorption, distribution, metabolism and toxicity. We intend to bring this strategy to Phase I first-in-human trials in the next year.
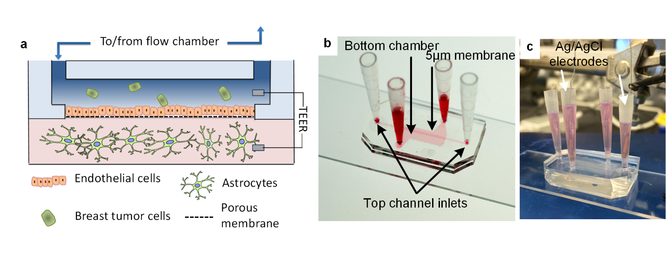
Metastasis from the primary tumor site to the brain is the most lethal complication of advanced cancer. 15% of breast cancers metastasize in the brain with a median survival of 5-14 months depending on the subtype. Therefore, it is critical to identify when a tumor has the clonal potential to metastasize to the brain. Current detection methods and treatment therapies have continued to improve but do not shed light on clonal metastatic potential, moreover, in vivo models do not recapitulate the complexity of the “live” micro-environment. A focus of our group is to develop a microfluidic device that mimics the cellular and physical components of the human blood-brain niche to study the brain metastatic process.
The device is composed of two chambers separated by a porous membrane. The top chamber and apical side of the membrane are seeded with human brain endothelial cells and use flow to mimic shear stress encountered within the vasculature. Cancer cells are introduced into this chamber in which they adhere to and migrate through the endothelium into the bottom chamber. The bottom chamber contains astrocytes suspended in a collagen gel to mimic the brain stroma and provide room for invading cancer cells to colonize and grow. Barrier integrity is monitored using TEER (trans-endothelial electrical resistance), and fluctuates as the tight junctions of the endothelium are compromised by invading cancer cells. This is characterized by IF and tight junction staining. Throughout all time points, from introduction into the flow chamber, adherence to the endothelium, extravasation through the barrier, migration into the stroma, and proliferation the cancer cells can be monitored via both microscopy and TEER.
Dr. Merajver's lab is applying this microfluidic blood-brain niche model to compare subclones of breast cancer cell lines in terms of their ability to extravasate, migrate and survive in the niche. We are using this information to characterize their migratory behavior from live-cell microscopy and comparative studies of metastatic markers for µBBN traversing and non-traversing cells when appropriate. We expect this will advance our ability to identify specific cells within heterogeneous populations that have a higher metastatic potential.
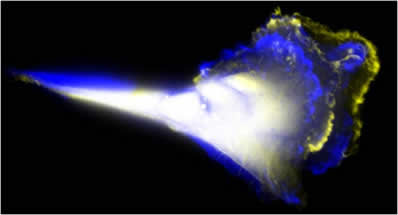
Time lapse of cancer cell invasion in a MDA-MB-231 tumoroid. Cells were seeded into the device for 24 hrs and invasion was further tracked for an additional 24 hrs.
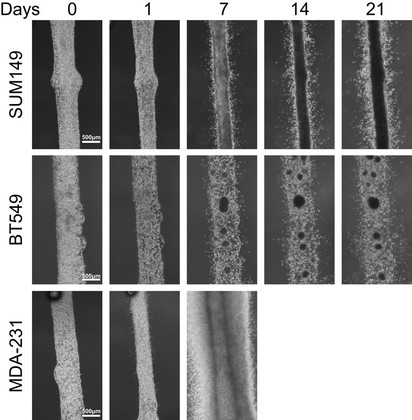
The study of invading leader cells at the tumor invasion front is of interest to our group as they may be guided by a targetable molecular phenotype which can be used to identify novel biomarkers for prognosis or therapy. In our lab, we have adopted the strategy of coupling functional phenotypic assessment of cell populations with molecular analysis as a directed approach to tease out the variations in cancer cells arising from tumor heterogeneity. Using a fluidic device for long-term (several days to weeks) 3-dimensional tumoroid culture of cancer cells, we have been able to recapitulate the dynamics of a tumor mass, the tumor invasion front and quantify the invasive potential of different triple negative breast cancer cell line models. Preliminary molecular analyses of the tumor invasion front using this device indicated a region of higher proliferation and suggest that the invading cells possess a different molecular phenotype from the tumoroid mass. The culture of cancer cells as tumoroids using this device presents a promising solution to the challenges posed by tumor heterogeneity when developing personalized therapeutics. We are actively pursuing the application of the device for both the quantification of disease progression risk, as well as the in-depth molecular characterization of the invasive subpopulations from patient samples and their response to tailored therapies.
Tumoroid culture of three triple negative breast cancer cell lines, SUM149, BT549 and MDA-MB-231. Tumoroid formation and invasion can be observed in these three cell lines over time, with invasion noted as early as 1 day after seeding the cells into the device.
Triple negative breast cancer (TNBC), one of the most aggressive breast cancer subtypes, carries a guarded prognosis and is the only subtype for which there are no approved targeted therapies. Chemotherapy is the mainstay of treatment both in the neo- and adjuvant settings and for metastatic disease. Improving patient outcomes requires a better understanding of the genotype and phenotypes of cells that mediate aggressiveness and metastases.
In the US, the incidence of TNBC is highest in women with African ancestry (AA); in western sub-Saharan Africa, single-institution studies show that TNBC constitutes 40- 80% of all breast cancers. Given the Caucasian/AA survival disparity in breast cancer, there is an urgent need to find actionable targets in TNBC of all ethnicities, but especially in TNBC in AA, which are suspected to be more aggressive. There is the urgent need to better understand the biology of TNBC in order to develop effective therapies targeting genes and pathways responsible for metastatic behaviors.
Breast cancer stem cells, the small population of cells in breast tumors that have been shown to mediate breast tumor initiation, metastasis, and resistance to conventional therapy have also been reported to mediate the heterogeneity of TNBC and are especially abundant in TNBC in women with African ancestry.
In this project, we seek to find differentially expressed and activated genes and pathways in the stem cell populations of patient derived TNBC xenografts from Ghanaian, African American, and Caucasian patients. By these analyses, we identified that the ALDH1 expressing cancer stem cells of these tumors segregate apart from bulk populations and CD24/44 stem cells, and were enriched with genes and pathways that are involved in tumor metastasis including the WNT, MAPK, and TGF-beta pathways. We also identified novel genes that are significantly up-regulated in this population that may serve as potential targets in further studies.
Petri dish studies in 2D have revolutionized our understanding of biology by enabling a reductionist approach. This approach simplifies experimental interpretation. We owe a great deal to those that have used these tools; however, the complexity of cancer requires models that are more biologically relevant. While the murine or mouse model is available, it is expensive due to the time investment necessary to care for and treat the animals. Therefore, we are exploring additive manufacturing as a means to produce breast ducts on demand to study breast cancer invasion while controlling the microenvironment around the cancer cells.
This lab has developed a three-dimensional printing technology that enables live cell printing at single cell resolution. In addition, we are developing the complimentary bio-material inks and software necessary to address this specific niche of 3D printing, through collaboration with engineers, material scientist and biologist specializing in cancer. Using what we learn we hope to develop an assay that can accelerate the rate of study of cancer formation, invasion, intravasation, and extravasation.
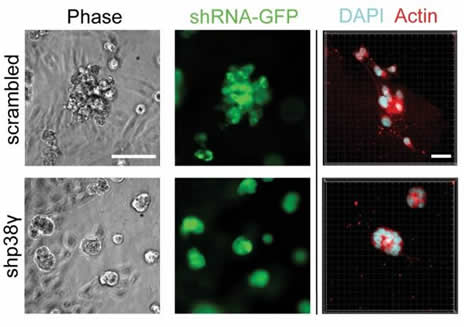
One hallmark of breast cancer progression is a shift from a slow epithelial mode of motion to a rapid mesenchymal-like form of motility. Understanding the genes and behaviors catalyzing this transition is therefore essential to targeting breast cancer metastasis. We discovered that a particular isoform of p38 MAPK, p38 gamma, regulates this motility transition by altering cytoskeletal architecture--in part due to regulation of RhoC expression. When p38 gamma is inhibited in aggressive breast cancer cells it dramatically impairs their ability to move, which appeared to be due to improperly aligned stress fibers. Significantly, high p38 gamma expression is associated with a worse patient prognosis, and inhibiting p38 gamma significantly restricts metastasis in a xenograft model.
Through collaborations with the Garikipati and Arruda labs in the Mechanical Engineering department we developed a computational model of cell motility that revealed that the improper stress fiber alignment was in fact responsible for the motility phenotype we observed. Surprisingly, the model predicted and subsequent biological experiments validated a novel leading edge behavior--leading edge protrusion oscillations--that allows cells to accommodate modified cytoskeletal architectures.
Future work will focus on dissecting the detailed molecular mechanisms by which p38 gamma influences breast cancer cell motility while concurrently expanding the capabilities of the computational model of motility.